The Semantic Measures Library Toolkit
The Semantic Measures Library Toolkit (SML-Toolkit) is a Java Toolkit dedicated to semantic measures computation and analysis. It is composed of various tools related to semantic measures computation and analysis. Those tools are provided through a common command-line interface.
The SML-Toolkit is developped in Java 1.7 and can therefore be used on any platform (Linux, Windows, Mac) in which Java is installed.
The latest version of the toolkit can be downloaded from the download section.
The main goal of the toolkit is to provide an easy-to-use toolkit to:
- Compute semantic measures scores using a wide range of measures.
- Evaluate semantic measures performance in a particular usage context, i.e. according to a specific benchmark.
- Generate/Evaluate benchmarks dedicated to specific usage contexts. Note that this functionality is only provided for particular usage contexts in which automatic generation of benchmarks is possible e.g. Gene Ontology study.
However, the toolkit currently focuses on semantic measure computation. It provides two command line interfaces for specifying a configuration which will be used to compute semantic measures:
- Domain-specific interfaces: The SML-Toolkit is a generic tool which can be used to compute semantic measures in a large variety of contexts (e.g. ontologies, measure configuration). However to ease its use, domain-specific interfaces (or profiles) have been developped in order to compute semantic measures in specific use cases, e.g. involving particular ontologies (the Gene Ontology, MesH)... Using a profile, i.e. a domain-specific interface, is the easiest way to use the toolkit. However not all usage contexts are taken into account.
- Generic configuration: A configuration can also be specified through an XML file (actually domain-specific interfaces generate such a configuration for you). This is a configuration mode for advanced users, it gives a low level access to the SML-Toolkit and therefore introduces more technical notions. This mode may therefore not suited for practitionners which know few about semantic measures and which do not have too much time to read the documentation.
- [Gene Ontology] - Easily compute semantic measures between Gene Ontology terms or gene products annotated by Gene Ontology terms
- [Gene Ontology] - Advanced use of the SML-Tooolkit to compare genes through their Gene Ontology annotations
Domain-specific interfaces
Profiles provide easy-to-use command-line interfaces for specific contexts of use, e.g. to compute semantic similarity between genes (products) annotated by Gene Ontology terms. Indeed, the generic configuration which can be made through the XML interface requires complex configuration files to be specified and is therefore not suited for most practionners.
To ease the use of the SML-Toolkit, specific profiles (or command-line interfaces) have been developped to wrap the generic configuration mode into domain specific layers. Those profiles aim at generating XML configuration files from command-line parameters. Such a configuration mode is therefore particularly adapted for users which are not experts of semantic measures and/or those only interested on a easy-to-use command line tool (not a geeky and complex tool ;).
Several domain-specific interfaces (or profiles) are supported by the SML-Toolkit. Below, the list of contexts of use for which a profile is/will be provided:
Graph-Based Semantic Similarity or Relatedness
- Gene Ontology: this profile can be used to compute semantic measure scores between Gene Ontology terms or gene products annotated by Gene Ontology terms.
- MeSH, the profile dedicated to the Medical Subject Heading. It can be used to compute semantic similarities between subject headings.
Semantic measures over the Gene Ontology
This profile is dedicated to the Gene Ontology (GO). It can be used to compute semantic measures scores between GO terms or gene products annotated by GO terms. The Gene Ontology must be in OBO format (version 1.2), the gene products annotations must be in GAF2 format or TSV (Tabulated Separated Values, an example is provided below). The Gene Ontology and associated annotations can be downloaded at http://www.geneontology.org.
To access the command-line interface dedicated to the Gene Ontology, type:Below the parameters which can be used, command-line examples are also provided:
Parameters
-go <file path>the path to the GO in OBO 1.2 format, other format are not supported (required). This file can be downloaded at http://www.geneontology.org/-goformat <format>The ontology file format, , accepted values [OBO, OWL, RDF_XML], default OBO-annots <file path>the path to the annotation file. Required for groupwise measures (-mtype gsee below) or any measure relying on a extrinsic metric (e.g. Resnik's Information Content) This file can be downloaded at http://www.geneontology.org/-annotsFormat <format>the format of the annotation file, accepted values [GAF2, TSV], more information about the TSV format above, default GAF2-queries <file path>the path to the file containing the queries. This file must contain the pairs of GO term or gene product ids separated by tabs (required). When similarities between gene products are computed, the ids are the values specified in the second column of the GAF2 file specifying the annotations of the gene products. An example is provided above.-output <file path>output file in which the results will be flushed (required).-
-mtype <type>the type of semantic measures you want to use:-
p(pairwise) to compute semantic measures between GO terms. -
g(groupwise) to compute semantic measures between gene products. Accepted values [p,g], defaultp. example-mtype p.
-
-
-aspect <flag>specifies the aspect of the GO to use:-
MF- Molecular Function -
BP- Biological Process -
CC- Cellular Component -
GLOBAL- the three aspects MF-BP-CC will be used using a virtual root between the three
When a groupwise measure is used, all gene products' annotations not related to the aspect selected will not be considered. Accepted values [MF,BP,CC,GLOBAL], defaultBP. example-aspect MF.
-
-
-notfound <flag>defines the behavior of the program if an entry element of the query file cannot be found, (i) in pairwise measures: one of the two GO terms cannot be found, (ii) in groupwise measures: one of the two gene products cannot be found. Accepted values [exclude, stop, set=]:-
excludethe entry will not be processed (a message will be logged if-quietis not used) -
stopthe program will stop -
set=<numerical value>the entry will not be processed and the given value will be set as score (a message will be logged if-quietis not used) default value = 'exclude'. Note that Gene products which are not annotated by at least one GO term of the selected aspect is removed and will therefore not be found despite it was defined in the annotation file.
-
-
-noannots <flag>defines the behavior if a gene product of the query file doesn't have any annotation (GO terms). Accepted values [exclude,stop, set=]:-
excludethe entry will not be processed (a message will be logged if -quiet is not used) -
stopthe program will stop -
set=<numerical value>the entry will not be processed and the given value will be set as score (a message will be logged if -quiet is not used) default value = 'set=-1' the score is set to -1.
-
-
-filter <params>this parameter can be used to filter the GO terms associated to a gene product when the provided annotation file is in GAF2 format.-
noEC=<evidence_codes>evidence codes to exclude separated by commas e.g.EC=IEAIEA annotations will not be considered -
Taxon=<taxon_ids>taxon ids separated by commas e.g.Taxon=9696to only consider annotations associated to Taxon 9696 Use:as separator betweennoECandTaxonif any is required. Example of value-filter noEC=IEA:Taxon=9696,5454. Default value no filter.
-
-pm <flag>a String value defining the pairwise measure to use. See the list of available measures here (required for pairwise measures or indirect groupwise measures).-gm <flag>a String value defining the direct groupwise measure or aggregation method (if an indirect groupwise measure must be used). See the list of available measures here (required for groupwise measures).-ic <flag>a String value defining the Information Content method to use (required by some measures). See the list of IC available here.-quietdo not show warning messages-notrgodo not perform a transitive reduction of the GO-notrannotsdo not remove annotation redundancies i.e. if a gene product is annotated by two GO terms {X,Y} and X is subsumed by Y in the GO, the GO term Y will be removed from the annotations.-threadsInteger defining the number of threads to use, i.e. processes allocates to the execution, default 1. Setting more threads reduce the execution time on large processes, suited configuration depends on your computer configuration, use with care if you don't get the implications in term of computational resources which will be used. Note also that results will not be ordered according to query file ordering.
Examples
Semantic similarity between Gene Ontology terms.
We compute the semantic similarity of each pair of GO terms specified in the query file -queries.
Those GO terms come from the Molecular function aspect of the Gene Ontology -aspect MF.
We use the measure proposed by Sclicker et al. and the Information content defined by Sanchez et al. (See the list of measures for references).
java -jar sml-toolkit-<version>.jar -t sm -profile GO
-go /data/go/eval/gene_ontology.1_2.obo
-mtype p
-queries /data/go/eval/input_query.tsv
-output /tmp/test-sml.tsv
-pm schlicker
-ic sanchez
-aspect MF
Example Download the following files (right-click on the link and save as):
- Last sml-toolkit version (available here)
- the ontology file go.obo (OBO Format) (downloaded from Gene Ontology website )
- input_query.tsv file :
GO:0000016 GO:0000016
GO:0000023 GO:0000023
GO:0000016 GO:0000023
http://purl.obolibrary.org/obo/GO_0000016 http://purl.obolibrary.org/obo/GO_0000016
http://purl.obolibrary.org/obo/GO_0000023 http://purl.obolibrary.org/obo/GO_0000023
http://purl.obolibrary.org/obo/GO_0000016 http://purl.obolibrary.org/obo/GO_0000023
java -jar ~/tools/sml-toolkit-0.9.4c.jar -t sm -profile GO
-go /data/go/go.obo
-mtype p
-queries /tmp/input_query.tsv
-output /tmp/test-sml.tsv
-pm schlicker
-ic sanchez
-aspect GLOBAL
Semantic similarity between Gene products.
We compute the semantic similarity of each pair of gene products identifiers specified in the query file -queries.
The annotations are in GAF2 format. We exclude the annotations inferred electronically (IEA) -filter noEC=IEA
We only want to consider the Biological Process aspect of the Gene Ontology -aspect BP.
We use the measure proposed by Lin et al., the Information content defined by Resnik and the Best Match Average miwing strategy - see the list of measures for more information.
If an entry contains no BP GO terms as annotations, it will not be found, we therefore set -1 as score --notfound set=-1.
Finally, we do not want the warnings to be shown -quiet and we allocate multiple threads to the process to reduce execution time -treads 4 (use with care, i.e. -treads 1, if you don't understand the implications).
java -jar sml-toolkit-<version>.jar -t sm -profile GO
-annots /data/go/goa_human.gaf
-filter noEC=IEA
-notfound set=-1
-go /data/go/go.obo
-mtype g -queries /tmp/input_query.tsv
-output /tmp/test-sml.tsv
-pm lin
-ic resnik
-gm bma
-aspect BP
-quiet -threads 4
Example Download the following files (right-click on the link and save as):
- Last sml-toolkit version (available here)
- the ontology file go.obo (OBO Format) (downloaded from Gene Ontology website )
- the annotations goa_human.gaf.gz (GAF Format) (save and extract the file, downloaded from Gene Ontology website )
- input_query_genes.tsv file :
P04637 O75056
O75056 O75056
P04637 P04637
java -jar sml-toolkit-<version>.jar -t sm -profile GO
-annots /data/go/goa_human.gaf
-filter noEC=IEA
-notfound set=-1
-go /data/go/go.obo
-mtype g -queries /tmp/input_query_genes.tsv
-output /tmp/test-sml.tsv
-pm lin
-ic resnik
-gm bma
-aspect BP
-quiet -threads 4
Execute the following command line (by specifying correct locations and replacing version id)
Extra documentation
Query file
One line per entry. An entry defines the two elements to compare (i.e. GO terms or gene products). use a unique tab to separate the two ids.
GENE_ID_1 GENE_ID_2
GENE_ID_2 GENE_ID_3
GENE_ID_4 GENE_ID_1
...
GO_TERM1 GO_TERM2
GO_TERM3 GO_TERM2
GO_TERM4 GO_TERM2
...
Annotation in TSV format
One line per gene product, use a unique tab to separate the gene product id and the GO terms ids. Two GO terms ids must be separated using a unique semicolon
GENE_ID_1 GO_TERM1;GO_TERM2;GO_TERM3
GENE_ID_2 GO_TERM1;GO_TERM3
...
MeSH: Semantic Measures
This profile is dedicated to the Medical Subject Headings (MeSH). It can be used to compute semantic measures scores between MeSH Descriptors or sets of MeSH Descriptors (e.g. documents annotated by MeSH descriptors). The MeSH must be in XML (2014 version supported), the sets of MeSH Descriptors must be in TSV format (Tabulated Separated Values, an example is provided below). The MeSH can be downloaded at http://www.nlm.nih.gov/mesh.
To access the command-line interface dedicated to the MeSH, type:Below the parameters which can be used, command-line examples are also provided:
Parameters
-mesh <file path>the path to the MeSH in XML format, other format are not supported (required). This file can be downloaded at http://www.nlm.nih.gov/mesh-annots <file path>the path to the annotation file. Required for groupwise measures (-mtype gsee above) or any measure relying on a extrinsic metric (e.g. Resnik's Information Content). This file is expected to be in XML format.-queries <file path>the path to the file containing the queries. This file must contain the pairs of MeSH Descriptors or sets ids separated by tabs (required).-output <file path>output file in which the results will be flushed (required).-
-mtype <type>the type of semantic measures you want to use:-
p(pairwise) to compute semantic measures between two MeSH descriptors. -
g(groupwise) to compute semantic measures between two sets of MeSH descriptors. Accepted values [p,g], defaultp. example-mtype p.
-
-
-notfound <flag>defines the behavior of the program if an entry element of the query file cannot be found, (i) in pairwise measures: one of the two MeSH descriptors cannot be found, (ii) in groupwise measures: one of the sets ids cannot be found. Accepted values [exclude, stop, set=]:-
excludethe entry will not be processed (a message will be logged if-quietis not used) -
stopthe program will stop -
set=<numerical value>the entry will not be processed and the given value will be set as score (a message will be logged if-quietis not used) default value = 'exclude'.
-
-pm <flag>a String value defining the pairwise measure to use. See the list of available measures here (required for pairwise measures or indirect groupwise measures).-gm <flag>a String value defining the direct groupwise measure or aggregation method (if an indirect groupwise measure must be used). See the list of available measures here (required for groupwise measures).-ic <flag>a String value defining the Information Content method to use (required by some measures). See the list of IC available here.-quietdo not show warning messages-notrdo not perform a transitive reduction of the MeSH-notrannotsdo not remove annotation redundancies i.e. if a set is annotated by two MeSH Descriptors {X,Y} and X is subsumed by Y in the MeSH, the MeSH descriptor Y will be removed from the annotations.-threadsInteger defining the number of threads to use, i.e. processes allocates to the execution, default 1. Setting more threads reduce the execution time on large processes, suited configuration depends on your computer configuration, use with care if you don't get the implications in term of computational resources which will be used. Note also that results will not be ordered according to query file ordering.
List of measures and metrics available
More information regarding the measures and metrics which can be used can be found at the dedicated webpage or in this spreadsheet.
Information content (IC):
-
Intrinsic
sanchezzhouseco
-
Extrinsic (requires annotations)
resnik
Pairwise measures:
- IC-based (requires an IC to be defined)
-
resnik -
lin -
schlicker -
jc
-
Groupwise measures:
-
Direct
tontouilpleeali_and_deanegic
-
Indirect (requires a pairwise measure)
minmaxavgavgnormbmabmmmaxnorm
Generic configuration
In the following documentation we present how the SML-Toolkit can be used to compute semantic measure scores according to an XML configuration.
The documentation first introduces the reader to general information related to the SML-Toolkit e.g. general configuration, knowledge base loading.
In a second part, the documentation covers the various tools provided by the toolkit.
A set of tutorials are also provided to easily take advantage of the toolkit in order to compute semantic measure scores.
Examples involving the Gene Ontology, MeSH, SNOMED-CT, OBO ontologies or RDF(S) graphs are also provided.
Consult the tutorial section for examples of use.
The documentation covers these topics:
- Generic configuration: General overview of the toolkit configuration
- Generic tags: Elements used in numerous tools
- Load Knowledge bases: How to define a knowledge base configuration
- XML tags: List of all XML tags
- Tools: The tools composing the SML-ToolKit
- SM: Fast computation of semantic measures' scores
- Notations
Profiles
Profiles are easy-to-use context specific command-line interfaces which can be used to compute semantic measures scores using the SML-Toolkit on particular context of use.
Profiles aim at hiding the complexity of using generic advanced configuration mode enabling to interact with the SML-Toolkit through XML configuration files used (presented below).
Thanks to those profiles the SML-Toolkit can be used specifying command-line parameters and associated values.
Notice that profiles are just wrappers of the generic XML interface and are only used to create an XML configuration file which will be processed fox by the SML-Toolkit engine.
Generic configuration
The tools provided by the SML-Toolkit are most of the time configured through an XML configuration file, this section covers the definition of such configuration files.
XML is a very intuitive markup language and SML-Toolkit configuration through XML doesn’t require being a computer scientist or expert in XML.
Users who have never eared about XML can be ensured that the following explanations will be sufficient to express a specific configuration.
The terminology defined here is adopted (see terminology, only tag and attribute have to be known).
The main tag or root tag of a configuration file must always be <sglib></sglib>.
The configuration will always be specified inside the sglib tag.
Allowed tags and attributes are defined by the documentation.
Thus, each tag must be a child of the unique <sglib> tag which must be specified in each configuration file.
However, for convenience, proposed examples will not always include the root tag.
An empty configuration file is presented below.
Note that comment can be added (surrounded by <!- and -->).
Generic tags
Numerous XML tags are generics as they are used by various tools e.g. tag enabling to specify the knowledge base configuration. We present those generic tags.
Definition of variables
Variables can be specified in order to facilitate the definition of the configuration file.
Variables can be useful to avoid recopying numerous time the same information e.g. workspace directory locations.
They will store a value which can therefore be used.
Variables must be defined in a unique <variables> tag and each variable must be specified through a <var> tag. To each <var> tag must be specified two attributes, the key and the value, respectively the name and the value associated to the variable. Variables with similar names are not allowed.
Variables can then be used in any attribute value of the XML configuration file using the special syntax {VARIABLE_KEY} e.g. {HOME} for a variable with key value HOME.
In this documentation we called {HOME} the pattern of the variable HOME.
At execution time, patterns will be replaced by the value associated to the variable they refer to i.e. in the example provided the variable {RESULT_REPO} will be associated to the value /tmp/sml/results.
A variable must be defined prior to its use in the variables tag variable definitions (top-down ordering).
A snippet of code used to define variables is presented below.
The variables are the first information taken into account by the configuration loader.
You can thus consider they can be used in all tags defined inside the root tag <sglib></sglib>.
Global options
Global options are defined as attributes of the tag <opt>.
The threads attribute allow you to specify the number of threads allocated to the process (~ the computation power allocated).
Define namespace prefixes
Depending on the specification and format in which is specified the knowledge base, namespace prefixes must sometimes be specified.
Namespaces can be viewed as containers in which a set of identifiers (e.g. URI) will be associated.
Namespace are useful to disambiguate identifiers.
In numerous cases, namespaces are complex strings and are thus replaced by prefixes.
As an example, the URI http://www.w3.org/2000/01/rdf-schema#Class will be replaced by rdfs:Class considering the namespace prefix rdfs associated to http://www.w3.org/2000/01/rdf-schema#.
Some knowledge base loader requires prefixes to be defined. For this purpose prefixes to consider can be specified in the namespaces tag.
Load Knowledge bases
Numerous tools rely on the definition of a knowledge base.
Graph-based knowledge bases are currently supported.
Their definitions are specified inside the graphs tag.
A semantic graph can be specified through the graph tag.
To each graph tag is associated an URI corresponding to the unique identifier of the graph.
This identifier will be used later on to refer to the graph.
A graph is loaded according to a dataset and actions to perform on the loaded data.
The loader will first load the dataset associated to the graph to then apply the actions associated to it.
Information related to both the data to load and the action to perform is detailed below.
Details concerning their XML configurations are also given.
Below is presented an example of knowledge base configuration.
Data
A graph can be loaded based on multiple data sources expressed in various formats e.g. RDF-XML, OBO.
Those files can easily be specified through the XML configuration file. Only local files are currently supported as data providers.
Depending on the data format considered a specific loader will be used.
Some loaders will expect some extra configuration parameters to be specified.
Please refer to corresponding loaders' specificities for detailed information.
Some of the supported formats are:
- OBO: Open Biomedical Ontology
- RDF-XML
- N-TRIPLES
- GAF-2: particular loader to load annotations expressed in GAF-2 format
Graph Loaders
This section provides information concerning the loaders currently supported.
OBO 1.2
Open Biological and Biomedical Ontologies (OBO) formatted ontologies can be specified as datasets.
In the following we adopt the terminology used in the official documentation.
Only objects associated to Stanza names Term and Type are currently loaded.
The OBO loader requires namespace prefix used in the ontology to be specified.
A default-namespace can also be specified, it will be used as default namespace for all non-prefixed Term and Type identifiers.
If no default-namespace is specified graph URI will be used as prefix.
The Default-namespace is specified using default-namespace attribute.
Terms are loaded as vertices and automatically typed as classes.
Is-a relationships are loaded as rdfs:subClassOf (According to the official OBO to OWL mapping ).
Object corresponding to Type Stanza name define the semantic relationships which can be used (and their properties).
Relationships defined in Term specifications are loaded as edges; the type of relationship is used as URI defining the semantic associated to the edge.
Alternative ids are currently not supported.
[Term] id: GO:0007379 name: segment specification namespace: biological_process def: "The process in which segments assume individual identities… is_a: GO:0007389 ! pattern specification process relationship: part_of GO:0035282 ! segmentation
The OBO definition above will be loaded as a graph composing of three vertices identified by the URIs GO:0007379, GO:0007389, GO:0035282 (with GO replaced by the defined GO namespace prefix).
A rdfs:subClassOf relationship will be created from GO:0007379 to GO:0007389, and a part_of relationship will be loaded from GO:0007379 to GO:0035282.
The part-of relationship will be associated to the URI default-namespace/part_of with default-namespace the value defined in the loader configuration.
Note that relationships used in term relationship definition require referring to a declared Type specification.
The current OBO loader fills minimal requirements in the context of semantic measures computation. Note however that a full and customizable OBO loader is under development.
RDF-XML
Statements are loaded considering subject and objects as graph vertices and predicate as relationship.
No types associated to vertices during loading.
N-TRIPLES
Statements are loaded considering subject and objects as graph vertices and predicate as relationship.
No types associated to vertices during loading.
GAF-2
The parser considers GAF 2.0 specification provided at:
- http://www.geneontology.org/GO.format.gaf-2_0.shtml
- http://wiki.geneontology.org/index.php/GO_Consortium_web_GAF2.0_documentation
Current loader loads the information as Graph elements.
The loader requires namespace prefixes used in the data file to be specified.
For each line:
-
DB_OBJECT_ID (column 1) is generated as a vertex typed as an instance and associated to an URI, (if the given vertex doesn’t exist). The URI is build considering the pattern prefix/DB_OBJECT_ID with prefix an optional prefix which can be specified as attribute and ID the value specified in the column. If no prefix is specified the graph URI is used as prefix. Use an empty prefix (i.e. prefix="") if none is required.
-
GO_ID (column 5) generates a vertex associated to a class (if the vertex doesn’t exist). The loader is aware of loaded prefixes.
-
A relationship DB_OBJECT_ID rdf:type GO_ID is created if qualifier column (col 4) is empty.
Filtering can be specified according to Evidence Code (column 7) and Taxons (column 12).
Xml configuration
A Data tag is used to specify the data to load into the graph.
Various specification format are currently supported e.g. rdf-xml, n-triples, obo.
A particular file is specified through the file tag in which two attributes must be given, the format and the path, respectively referring to the file format defining the loader which will handle the file and the location of the file.
Depending on the specified format a particular loader will be used to load the data. Refer to the technical documentation for in-deep explanations of the loader behaviour. Note that, depending on the loader selected, particular attributes must be required or used to specify additional parameters.
| Format | Value |
|---|---|
| OBO | obo |
| RDF-XML | rdf-xml |
| N-TRIPLES | n-triples |
| GAF2 | gaf_2 |
Actions
Actions are used to perform particular treatments to the knowledge base after data loading e.g. removal of particular nodes.
Actions are specified under the graph definition (inside the graph tag) through a tag called actions.
For each action an action tag is specified.
To each action is associated a type defining the action to perform.
Action execution order is defined by the ordering of their definition.
Particular attributes can be required or used to specify additional parameters.
In the following we detail the various actions currently supported. In order to apply a particular action the flag associated to the type of action must be specified as value of the attribute type.
Transitive Reduction
Perform a transitive reduction according to rdfs:subClassOf or rdf:type relationships i.e. remove all useless direct relationships according to the relationships redundancy definitions. In other terms if a class A is specified as a subclass of a class B, any superclass of B called C is also a superclass of A. All A rdfs:subClassOf C relationships must be removed as they are not expected by semantic measures. This property relies on the transitivity of the rdfs:subClassOf relationships. A transitivity over rdf:type relationships can also be performed in order to remove redundant annotations i.e. if an instance is annotated to classes A and B with A a subclass of B thus B can be inferred and must be removed.
XML configuration
Flag: TRANSITIVE_REDUCTION
The action expects an extra attribute specifying the type of target to consider. The name of the attribute is target. Admitted values are ‘CLASSES’ and ‘INSTANCES’ respectively denoting that the transitive reduction must be performed according to rdfs:SubClassOf or rdf:type relationships.
Example
Numerous semantic measures are affected by relationship or annotation redundancies. In the figure proposed below all edges rdfs:SubClassOf relationships. Red Dotted relationships can be removed without loss of information as they can be inferred from the others (black ones). Applying the action with target="CLASSES" will remove all redundant red edges.
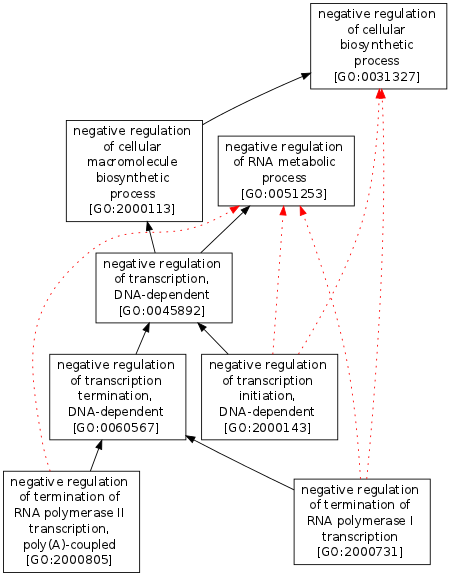
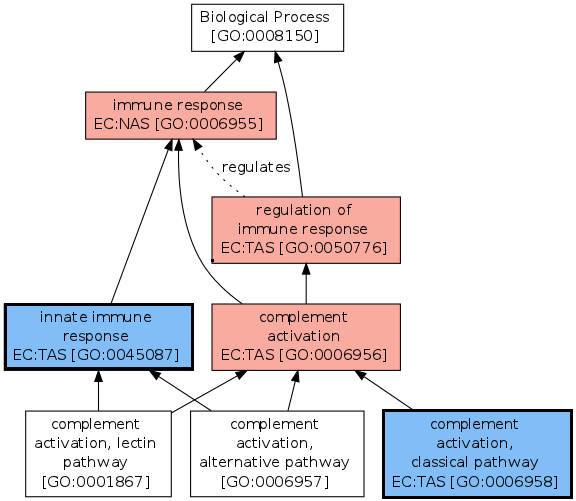
Redundant annotations can also be easily removed. In the figure presented below, colored vertices correspond to the annotations of the human gene P01773. Red vertices (i.e. annotations) can be inferred through the true path rule i.e. a gene is annotated by all nodes subsuming its direct annotations (blue vertices).
Vertices Reduction
Perform a reduction of the vertices according to a given pattern, vocabulary or traversal.
As an example, when a pattern is specified all vertices associated to an URI matching the pattern will be removed.
Related edges are also removed.
This action can also be used to remove the part of the semantic graph which is not related to the taxonomic graph induced by a specific concept.
As an example, consider a large semantic graph containing various aspects of knowledge, e.g. the Gene Ontology contains three specific aspects (Molecular Function, Cellular Component, and Biological Process) which are represented by a specific part of the ontology.
If you only want to consider the graph induced by a single aspect (e.g. Cellular component), you will have to remove the part of the ontology which is not related to this aspect.
The vertices reduction action can be used to perform such a process.
XML configuration
Flag: VERTICES_REDUCTION
Attribute ‘regex’ is used to specify a particular regex (Java syntax) to evaluate vertices’ URI.
Attribute ‘vocabulary’ is used to specify that vertices associated to a particular vocabulary have to be removed. The attribute cannot be used with regex attribute. Supported vocabularies are RDF, RDFS and OWL i.e. vocabulary=”RDF”. Use comma as separator if multiple vocabularies requires to be specified.
Attribute ‘file_uris‘ is used to specify the location of a file containing a bench of uris to remove. Please specify a unique URI per line. Multiple files can be specified using comma as separator.
Attribute ‘root_uri‘ is used to specify a root node typed as a CLASS. Only The Taxonomic graph induced by the specified root is considered, i.e. all the vertices of type CLASS which are subsumed by the root. Existing semantic relationships between vertices of type CLASS not removed are also considered. Then, only the instances typed by a conserved vertex of type CLASS will be part of the graph, i.e. the instances establishing a relationship rdf:type with a class composing the new underlying taxonomic graph.
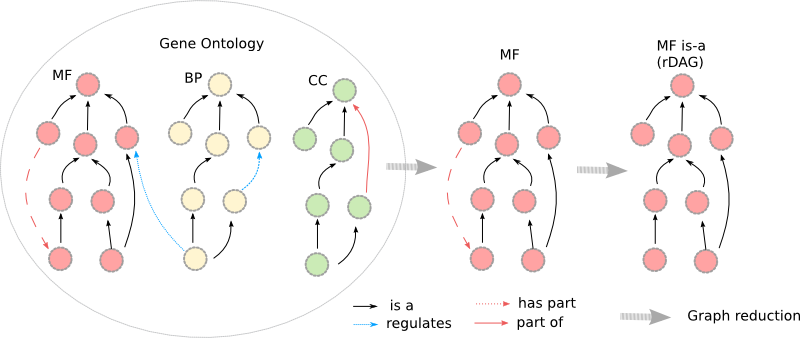
The Gene Ontology (GO) is composed of three orthogonal sub-ontologies, i.e. MF (Molecular Function), BP (Biological Process) and CC (Cellular Component); each of them:
- Can be isolated only considering relationships established between their respective terms.
- Can be reduced as a rooted Directed Acyclic Graph (rDAG) only considering taxonomic relationships.
- Contains a unique term subsuming the others (is rooted), GO:0003674 for MF, GO:0008150 for BP, GO:0005575 for CC.
- Forms a connex graph if we only consider taxonomic relationships.
- Is acyclic if we only consider "is-a" (and "part-of") relationships.
Numerous treatments based on semantic measures which are applied to the GO only consider a unique sub-ontology or require the considered graph to be rooted i.e. to contain a unique term subsuming all terms. In this cases, a root term can be specified (e.g root_uri="GO:0003674") in order to only consider the sub-ontology related to the Molecular function sub-ontology. Only the taxonomic graph induced by the set of terms composed of GO:0003674 (root) and the GO terms subsumed by it will be considered. Other vertices and relationships for which the source and the target are not linked to the reduced taxonomic graph are removed.
The graph which will be generated by the treatment is the one located in the middle of the figure proposed below.
Rerooting
The aim of the action is to root the semantic graph according to the given root and rdfs:subClassOf relationships.
The algorithm searches all the vertices which are typed by CLASS vertex type and without outgoing rdfs:subClassOf relationship (i.e. taxonomical relationship).
Those vertices are considered as roots.
The given vertex is added to the graph if it doesn't exist.
The roots are then considered as rooted by the specified new root, i.e. a rdfs:subClassOf relationships is created from each element of the root set to the new root.
XML configuration
Flag: REROOTING
An attribute ‘root_uri’ must be given as parameter in order to define which node must be considered as new root. The root node is specified through it URI. If the node doesn't exist it will be created, its associated vertex type will be CLASS. A special value __FICTIVE__ can also be used in order to root the graph using a fictive root rooting all nodes of type CLASS which do not contain an out relationships of type rdfs:subClassOf .
Example
XML Tags
For more information about the design details of the XML configuration file please consult the updated list of XML tags and attributes located at here. See embedded version below. Note that some tags are specific to particular tools and are therefore detailed in the following documentation.
Tools
SM: Computation of Semantic measures' scores
The SM tool is dedicated to semantic measures computation considering a particular XML configuration file defining (i) the knowledge base, (ii) the measures, (iii) the semantic elements on which the evaluation have to be made e.g. pair of concepts or instances URIs.
Numerous measures can be used; an updated list of semantic measures and related functions available can be consulted here (or see below).
Execution of the tool can easily be launched using:
java -jar sml-toolkit-<version>.jar -t sm -xmlconf xml_file
The definition of the XML configuration file is explained in the next section.
XML configuration
The XML configuration enables the definition of the knowledge base to consider, the measures to evaluate and the query to consider. The definition of a knowledge base configuration through an XML file has already been introduced. This section only provides information about specific configuration elements of the SM tool.
Configuration must be specified inside the sml tag.
Attribute module set to sm must also be specified.
The graph involved in the process also has to be specified using graph attribute in which the URI of the considered graph must be specified.
Information content
Information contents (ICs) are defined around a unique <ics> tag. ICs are specified using <ic> XML tag composed of attributes:
- id: an unique identifier used to refer to the IC. Duplicate IC identifiers are not allowed [required].
- flag: a literal referring the method used to compute the Information Content [required]. Available flags and corresponding computation methods can be found here .
- label: the label given to the specified IC [optional]. The id is used as label if no value is specified.
Two kinds of approaches can be distinguish to compute term's IC , intrinsic ones only using topological information (ICi) and corpus-based (ICc) taking advantage of concepts distribution in the knowledge base (number of occurrences i.e. instances annotated by the concept).
Pairwise measures
Pairwise measures are defined around a measures tag for which type attribute is specified to "pairwise”.
Pairwise measures are thus specified using attributes:
- id: an unique identifier used to refer to the measure. Duplicate identifiers are not allowed [required].
- flag: a literal referring the SM used [required]. Available flags and corresponding pairwise measures can be found here.
- label: the label given to the specified measure [optional]. The id is used as label if no value specified. The label will be used in the output file.
Some pairwise measures require an IC (dependencies are specified in the measure definition). If the selected flag is associated to such measure the chosen IC must be specified using ic attribute:
- ic: an IC id mapping to an IC definition [required if an IC-based measure flag is used].
Note that some semantic measures admit or require extra parameters.
Advanced: the value [FULL_LIST_IC] can be used as value of the ic tag in order to apply all possible configuration. The flag of the measures will be generated considering [flag]_[ic_flag] pattern (remind that the id is used as pattern if no flag is explicitly specified).
Groupwise measures
Groupwise measures are defined around a measures tag for which type attribute is specified to "groupwise". All measures are specified using measure tag specifying attributes:
- id: a unique identifier used to refer to the measure. Duplicate identifiers are not allowed [required].
- flag: a literal referring to the semantic measure used [required]. Available measures and corresponding flags can be found here.
- label: the label given to the specified measure [optional]. Duplicate labels are not allowed, if no label is specified the id will be used as label. The label will be used in the output file.
Two kinds of groupwise measures can be distinguished:
- Mixing strategies; groupwise measures relying on a pairwise measure to compute annotations similarity/relatedness to then aggregate the scores. Those measures require a pairwise measure to be configured (see above for pairwise measure definition).
- Standalone; these measures do not rely on a pairwise measure.
Mixing strategy based groupwise measures also called indirect groupwise measures require a pairwise measure to be defined. If the selected flag is associated to an indirect groupwise measure a pairwise measure must be specified through pairwise_measure attribute.
- pairwise_measure: a pairwise measure id mapping to pairwise measure definition [required if a mixing strategy measure flag is used].
Some groupwise measures require an IC (dependencies are specified in the measure definition). If the selected flag is associated to such measure the IC to use must be specified using ic attribute.
- ic: an IC id mapping to an IC definition [required if an IC-based measure flag is used].
Note that some semantic measures admit or require extra parameters e.g. IC. Extra-parameters are defined in the page dedicated to semantic measure methods description.
Advanced: You can use the value [FULL_LIST_PM] to the pairwise_measure attribute, all measures will be computed. In the output file indirect groupwise measure label will be build respecting the pattern [pairwise_measure_flag]_[groupwise_measure_flag].
Queries
Queries to perform can be defined using queries XML tag.
The tag can be used to define numerous semantic measures scores to compute for both pairs of concepts or entities annotated by concepts.
The input file must contain pair of semantic elements to evaluate.
The SM tool will compute the scores for all parameterized measures i.e.all pairwise measures when comparisons only involve concepts (classes) or all groupwise measures defined when the comparison involves entities.
Note that a unique measure configuration can be specified.
Attributes associated to the queries tag are:
- id: an unique identifier used to refer to the queries. Duplicate identifiers are not allowed [required]. The identifier is currently only used for logging.
- type: the type of queries specified, specify oTOo value (Object TO Object) for instance comparisons [required]. Allowed values are oTOo and cTOc respectively Object TO Object and concept TO concept comparison i.e. groupwise and pairwise.
- file: input file defining the comparison to make (see an example below) [required].
- output: output file in which scores will be printed [required].
- uri_prefix [optional] a prefix to add to all identifiers specified in the queries.
Example of XML configuration
Input file example
Output file example
e1 e2 MAX_dice BMM_dice NTO TO http://g/P62633 http://g/P07910 0.6 0.55 0.4 0.62 http://g/O00567 http://g/P01106 0.7 0.5 0.46 0.43 http://g/Q99816 http://g/Q08426 0.71 0.6 0.5 0.67 ...
Supported measures and metrics
Measures and metrics available are specified in a public Google document available here .
An embedded version is proposed below, use tabs to switch between different types of metrics.
Notations
Some of the notations used in this documentation are specified in W3C RDF documentation.
- rdfs:subClassOf refers to http://www.w3.org/2000/01/rdf-schema#subClassOf
- rdf:type refers to http://www.w3.org/1999/02/22-rdf-syntax-ns#type
